
Naukowcy z Meta AI udostępnili „model języka białkowego” ESM-2 z 15 miliardami parametrów oraz bazę ESM Metagenomic Atlas z ponad 600 milionami metagenomicznych struktur predykcyjnych.
Announcing the ESM Metagenomic Atlas — the first comprehensive view of the ‘dark matter’ of the protein universe. Made possible by ESMFold, a new breakthrough model for protein folding from Meta AI.
— Meta AI (@MetaAI) November 1, 2022
More in our new blog ➡️ https://t.co/LsUhSjzqCf
1/3 pic.twitter.com/5lq48rPv5A
Białka są złożonymi cząsteczkami składającymi się nawet z 20 rodzajów aminokwasów i pełnią w organizmach wszelkie funkcje biologiczne. Są one składane w złożone trójwymiarowe struktury, których kształt bezpośrednio wpływa na sposób ich działania.
Określenie rodzaju związku pozwala naukowcom zrozumieć, jak funkcjonują białka. Ponadto dane o kształcie pomagają im znaleźć sposoby naśladowania, modyfikowania lub przeciwdziałania tym zachowaniom.
Nie można wziąć wzorów aminokwasów i od razu określić ostatecznej struktury, a symulacje i eksperymenty są czasochłonne.
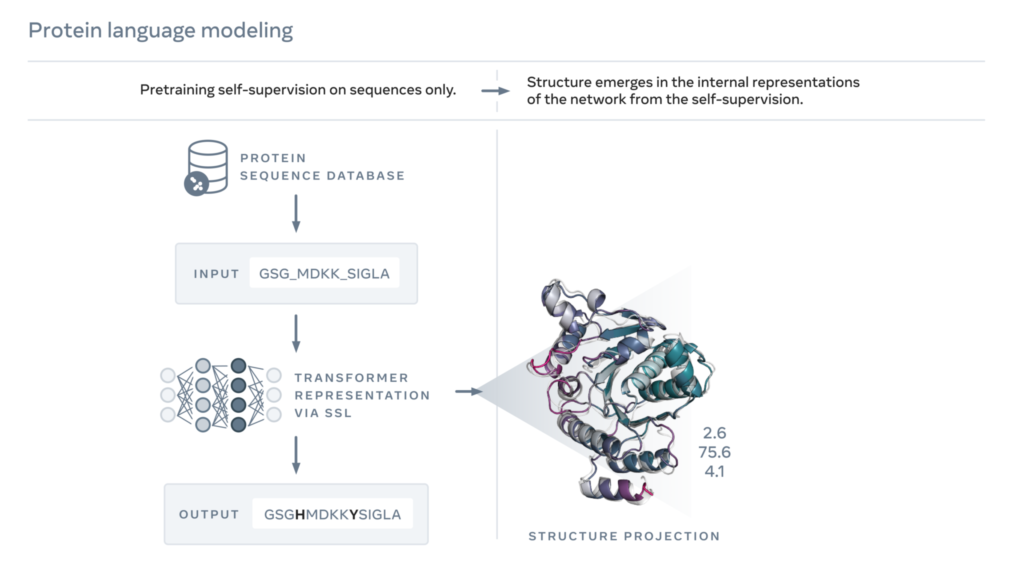
Meta AI powiedziała, że sieć neuronowa ESM-2 to duży model językowy zaprojektowany do „badania wzorców ewolucyjnych i tworzenia dokładnych przewidywań związków bezpośrednio z sekwencji białka”.
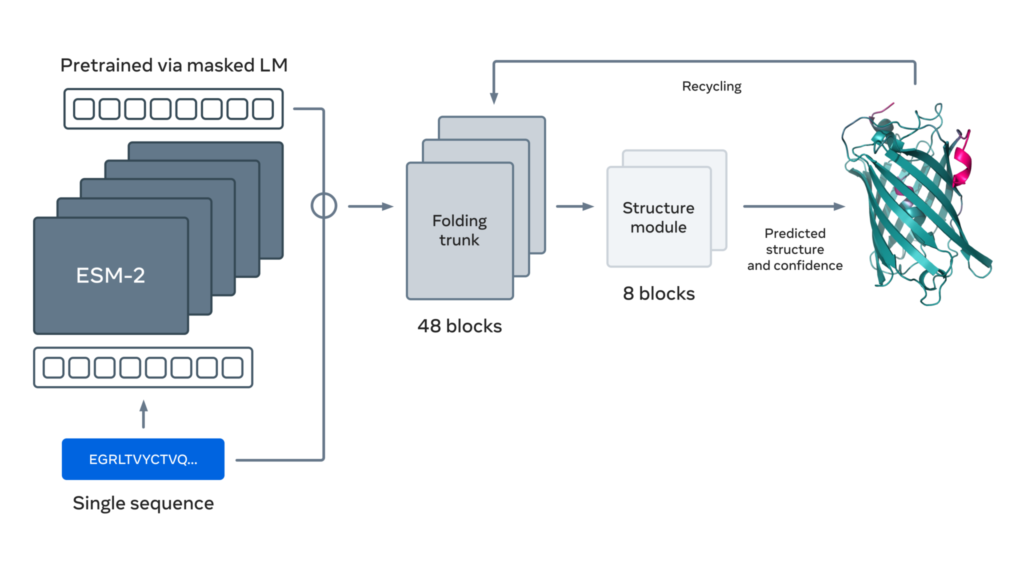
System przetwarza sekwencje genów wykorzystując technikę samouczenia zwaną maskowanym modelowaniem języka.
Naukowcy twierdzą, że wytrenowali algorytm na tablicy sekwencji milionów naturalnych białek. W dokumencie możemy przeczytać:
W tym podejściu model musi poprawnie uzupełnić słowa we fragmencie tekstu, np. 'To __ or not , that is ’. Wyszkoliliśmy model językowy, aby wypełnić luki w sekwencjach białek, takich jak 'GL_KKE_AHY_G’ wśród milionów różnych związków.
ESM-2 – co to?
ESM-2 jest największą i najbardziej wydajną siecią neuronową tego typu. Według naukowców algorytm jest 60 razy szybszy niż inne zaawansowane systemy, takie jak AlphaFold firmy DeepMind.
Algorytm pomógł zbudować Atlas Metagenomiczny ESM, przewidując 617 milionów struktur z bazy danych białek MGnify90 w ciągu zaledwie dwóch tygodni na klastrze złożonym z 2000 jednostek GPU. Symulacja związku składającego się z 384 aminokwasów zajęłaby 14,2 sekundy na jednej karcie graficznej Nvidia V100.
Przy dzisiejszych narzędziach obliczeniowych przewidywanie struktury setek milionów białek może zająć lata. Nawet przy wykorzystaniu zasobów dużej instytucji badawczej. Aby dokonywać predykcji w skali metagenomiki, przełom w szybkości przewidywania jest krytyczny.
Meta AI ma nadzieję, że ESM-2 i Atlas Metagenomiczny ESM przyczynią się do rozwoju nauki i pomogą ekspertom badającym historię ewolucji lub zwalczającym choroby i zmiany klimatyczne. Naukowcy dodają:
Badamy również sposoby zastosowania modeli językowych do opracowania nowych białek i pomocy w rozwiązywaniu problemów zdrowotnych i środowiskowych.
Przypomnijmy, że w lipcu algorytm AlphaFold firmy DeepMind przewidywał niemal każdy znany nauce związek występujący w roślinach, bakteriach i zwierzętach.
W tym samym miesiącu naukowcy z MIT opracowali EquiBind, model głębokiego uczenia, który łączy cząsteczki z białkami w tworzeniu leków 1200 razy szybciej niż jego odpowiedniki.
W lipcu 2021 roku sztuczna inteligencja z DeepMind wymodelowała 20 tysięcy struktur ludzkich białek.